- solen.sk - FIBROSE CÍSTICA (1. FIBROSE CÍSTICA (Parte 1))
- solen.sk - FIBROSE CÍSTICA (2. FIBROSE CÍSTICA (Parte 2))
- sazch.sk - FIBROSE CÍSTICA
- health.gov.sk - Novos medicamentos modernos para doentes com fibrose quística.
- clubcf.sk - Orkambi
- medicinesinfo.sk - Medicação Kaftrio
- mayoclinic.org - Fibrose cística
- cff.org - Fundação da Fibrose Cística
- medlineplus.gov - Visão geral da fibrose cística
O que é a fibrose quística e quais os seus sintomas? Causas, diagnóstico

Fonte fotográfica: Getty images
A fibrose quística é uma doença hereditária em que os canais de cloreto nas membranas celulares estão danificados. A doença é causada por uma mutação genética no gene da FC.
Sintomas mais comuns
- Mal-estar
- Dor abdominal
- Dor à volta do umbigo
- Dor durante as fezes
- Febre
- Cãibras no abdómen
- Espiritualidade
- Prisão de ventre
- Açúcar na urina
- Depressão - humor deprimido
- Diarreia
- Fezes com sangue - sangue nas fezes
- Hemorragia
- Pele azul
- Indigestão
- Náuseas
- Inchaço dos membros
- A ilha
- Dedos atarracados
- Nariz cheio
- Tosse seca
- Fraqueza muscular
- Comichão na pele
- Fadiga
- Ansiedade
- Tosse húmida
- Água no abdómen
- Vómitos de sangue
- Vómitos
- Tosse com muco
- Tosse com sangue
- Brancos dos olhos amarelos
- Pele amarelada
- Aumento do fígado
- Aumento dos níveis de açúcar no sangue
- Aumento da temperatura corporal
Mostrar mais sintomas ᐯ
Características
A fibrose quística (FC) é uma doença hereditária que afecta vários órgãos do corpo humano, sendo reconhecível à nascença, mas algumas formas podem não ser diagnosticadas até à idade adulta.
Pensa-se que a parte mais grave da fibrose quística é a lesão dos pulmões e do fígado, que é, em última análise, a causa da redução da vida dos doentes com fibrose quística.
O tratamento é complexo e incide sobre os sintomas em todos os sistemas de órgãos. O insucesso do tratamento conservador é seguido de um transplante de órgãos, que é frequentemente necessário numa idade jovem.
A fibrose quística é uma doença conhecida desde a Idade Média, cujo principal sintoma é a produção de muco, razão pela qual lhe foi dado o nome mais antigo - mucoviscidose.
A primeira descrição médica da doença foi feita em 1936 pelo Dr. Fanconi, um pediatra que trabalhava na Suíça, ao qual se juntaram outros médicos e cientistas que acrescentaram importantes descobertas ao diagnóstico.
Uma das descobertas mais importantes em relação a esta doença foi a localização do gene cuja mutação causa a fibrose quística e os seus sintomas.
Esta descoberta foi efectuada pelos biólogos moleculares Robert Williamson e L. C. Tsui, que localizaram o gene em 1985 e o isolaram com sucesso, bem como o seu produto, em 1989.
O gene da fibrose quística está localizado no cromossoma 7 e o seu produto é o regulador de condutância transmembranar da fibrose quística (CFTR).
Trata-se de um canal de cloreto que se encontra nas membranas de todas as células do corpo humano e que passa os iões de cloreto para fora ou para dentro da célula. As mutações neste gene fazem com que o canal de cloreto se torne disfuncional.
Uma em cada 2.500 crianças nasce com uma mutação no gene CFTR. Esta é a doença hereditária mais comum na raça caucasiana. No passado, a doença era considerada fatal porque as crianças frequentemente não atingiam a puberdade.
Atualmente, com os tratamentos modernos, a fibrose quística é considerada uma doença que "encurta a vida".
Uma pessoa com fibrose cística pode viver até aos 45 anos de idade.
No entanto, o transporte da mutação CFTR é muito mais comum na população. Aproximadamente uma em cada 25 pessoas é portadora da mutação do canal de cloreto.
Paradoxalmente, na Idade Média, estas pessoas eram favorecidas com "imunidade" à cólera mortal (o agente causador do Vibrio cholerae).
A cólera provocava uma diarreia maciça, com uma fuga maciça de sais e de água do corpo, e as pessoas morriam sobretudo devido à desidratação e à perturbação do ambiente interno.
No entanto, a toxina da cólera não se podia ligar ao canal CFTR mutado, pelo que as pessoas com esta mutação não desenvolviam diarreia com fuga de cloreto e água.
Compromissos
A principal causa da fibrose quística é uma mutação no gene CFTR (Cystic Fibrosis Transmembrane Conductance Regulator), que está localizado no braço longo do cromossoma 7.
Na prática, isto significa que ambos os pais são portadores da mutação, chamados heterozigotos assintomáticos (Aa).
Se o seu filho herdar o alelo recessivo (a) de cada um dos pais, a criança desenvolverá a doença (aa). Se ambos os pais forem portadores do gene mutado, a probabilidade de ter um filho doente é de 25%.
A doença é sempre herdada horizontalmente, ou seja, de pai para filho, em duas gerações. Ambos os sexos podem ser igualmente afectados. Nos casamentos consanguíneos, o risco da mutação aumenta.
Existem atualmente cerca de 1500 mutações conhecidas que podem danificar o gene da fibrose quística e desativar o canal de cloreto.
A disfunção do gene é definida como uma falha completa do funcionamento do gene, uma incapacidade de integração na membrana celular no local correto ou uma produção insuficiente do gene no organismo.
Para além de mover os iões cloreto para dentro e para fora da célula, o canal de cloreto tem muitas outras funções.
Regula outros canais de membrana para o movimento de outros iões, acidifica os organelos celulares (especificamente o aparelho de Golgi, que está ativamente envolvido no transporte de produtos importantes), regula o transporte de proteínas para dentro e para fora da célula e é responsável pela capacidade antibacteriana e bactericida das células através da formação de óxidos de azoto.
A disfunção dos canais de cloreto provoca níveis elevados de iões cloreto (Cl) e sódio (Na) no suor, pelo que o suor das crianças com fibrose quística é salgado (NaCl).
Há um aumento da absorção de sódio noutros sistemas de órgãos importantes, como o sistema respiratório, o sistema gastrointestinal (especialmente o pâncreas, a vesícula biliar, o fígado e o intestino) e o sistema reprodutor.
Uma vez que o sódio também é absorvido pelas células a uma velocidade superior à da água, todas as secreções produzidas pelos órgãos ficam desidratadas.
A desidratação das secreções resulta em muco espesso e viscoso no trato respiratório, diminuição da secreção de enzimas pancreáticas, bílis espessa, muco espesso nos intestinos e no trato genital e diminuição da produção de esperma.
A acidificação deficiente (diminuição do pH) do aparelho de Golgi resulta numa diminuição da produção de importantes compostos químicos de enxofre que têm a função de "diluir" o muco.
Uma segunda função importante do aparelho de Golgi é a atividade das sialotransferases, cuja perturbação resulta na produção de asialoglicosídeos, que subsequentemente formam um recetor para bactérias, principalmente do grupo de espécies Pseudomonas.
Trata-se de estirpes de bactérias resistentes a uma vasta gama de antibióticos, cujo tratamento é difícil e moroso. A colonização crónica por este agente patogénico é típica dos doentes com fibrose quística.
Sintomas
A fibrose quística é uma doença que afecta múltiplos órgãos e a disfunção dos canais de cloreto afecta todos os órgãos com a sua perda progressiva.
Sintomas do sistema respiratório
O muco espesso presente nas vias respiratórias obstrui especialmente as pequenas partes da árvore brônquica, impedindo também o movimento dos minúsculos cílios, que são finas projecções da membrana mucosa que desobstruem as vias respiratórias através do seu movimento ondulante.
Além disso, o ar expirado contém níveis baixos de azoto, que tem uma função antimicrobiana.
Estes processos patológicos resultam nos seguintes sintomas:
- inflamação crónica imunomediada das vias respiratórias
- bronquiectasias (dilatação dos bronquíolos com inflamação)
- formação de quistos com um rebordo de tecido fibroso (rígido e inflexível)
- inflamação crónica dos seios paranasais - pansinusite
- pólipos nasais
- infecções repetidas com bactérias resistentes dos géneros Pseudomonas, Streptococcus Aureus, Haemophilus Influenzae e Burkholderia Cepacia, que conduzem à síndrome de cepacia (inflamação dos pulmões com formação de abcessos e desenvolvimento de sépsis e falência de órgãos)
- micoses frequentes - infecções do trato respiratório por fungos
- insuficiência respiratória (falência pulmonar) que leva à morte prematura
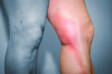
- dedos rígidos - causados pela abertura das ligações vasculares durante uma hipoxia prolongada (fornecimento insuficiente de oxigénio aos tecidos)
Sintomas do sistema digestivo
O muco espesso presente nos intestinos impede o movimento das vilosidades intestinais e prejudica a passagem intestinal. Ocorre um bloqueio intestinal.
Este sintoma pode estar presente num bebé já no útero. O mecónio (semelhante às fezes fetais) acumula-se nos intestinos e o bebé não consegue expelir as primeiras fezes após o nascimento. Desenvolve-se uma obstrução intestinal chamada íleo meconial.
Na idade adulta, esta situação designa-se por síndrome de obstrução intestinal distal (SIOD) e é causada pela ingestão insuficiente de líquidos ou pela ingestão de alimentos pegajosos, como o milho cozinhado.
O pâncreas é uma glândula que segrega sucos pancreáticos líquidos que contêm enzimas digestivas. Quando estes sucos se tornam espessos e escorrem, os ductos da glândula ficam bloqueados.
No entanto, o pâncreas não pára de produzir esses sucos, que se acumulam no interior do órgão e formam quistos, que são envolvidos por tecido fibrótico duro.
A pressão da secreção acumulada e os numerosos quistos degradam o tecido pancreático, que perde gradualmente a sua função de produção de enzimas.
Os quistos cheios de suco rebentam quando estão demasiado cheios. As enzimas digestivas derramam-se nos órgãos circundantes, digerindo, destruindo e "mordiscando" os órgãos abdominais circundantes.
Esta situação é acompanhada de dores fortes na parte superior do abdómen, à volta do umbigo e por baixo da caixa torácica esquerda, a que se dá o nome de pancreatite.
A pancreatite repete-se episodicamente. O tecido inflamado cicatriza e forma uma cicatriz fibrótica. Desenvolve-se a fibrose quística do pâncreas. Este sintoma típico pode ser o primeiro e único sinal de fibrose quística.
A inflamação repetida do pâncreas e a formação da cicatriz fibrosa também danificam os ilhéus de Langerhans, que produzem insulina. A redução da secreção de insulina no sangue resulta no desenvolvimento de diabetes mellitus.
Os canais biliares contêm bílis espessada, que é difícil de excretar. Ocorre a estase da bílis e a formação de cálculos biliares. A inflamação crónica leva à necrose (morte) das células e à formação de tecido fibrótico, o que conduz à cirrose hepática.
As defesas reduzidas da membrana mucosa do trato digestivo levam à proliferação de bactérias, especialmente clostrídios, que causam diarreia, que é acompanhada por distúrbios pancreáticos com um elevado teor de fezes gordurosas.
Resumo dos sintomas do TGI:
- obstrução intestinal
- pancreatite recorrente
- diabetes
- formação de cálculos biliares e inflamação da vesícula biliar
- cirrose hepática
- diarreia clostridial
- prolapso do reto
Desnutrição em doentes com FC
Os doentes com fibrose quística, que causa dificuldades respiratórias, tosse crónica e infecções recorrentes, têm necessidades nutricionais acrescidas.
A má passagem intestinal, a absorção prejudicada de nutrientes através do muco espesso que cobre a mucosa intestinal, a disfunção do pâncreas e a redução das enzimas digestivas, os cálculos biliares e a secreção prejudicada de bílis no intestino causam uma digestão e absorção prejudicadas de nutrientes importantes, especialmente gorduras.
Além disso, a deglutição de muco tossido, as dores abdominais e a presença de inflamação crónica no organismo reduzem o apetite. Os doentes com FC sofrem, por isso, de subnutrição e desnutrição, podendo sofrer de carências vitamínicas graves, sobretudo das vitaminas lipossolúveis, ou seja, A, D, E e K.
Sintomas no sistema reprodutor
A maturação sexual e o início da puberdade são atrasados nas crianças com FC, devido à inflamação crónica e à desnutrição, principalmente devido à falta de gorduras, que são essenciais para a maturação sexual e para a produção de hormonas sexuais.
A maioria dos homens (98%) com fibrose quística são inférteis, o que se deve ao estreitamento ou "colagem" das paredes de ambas as trompas de Falópio.
As trompas de Falópio transportam os espermatozóides dos testículos para a uretra, onde são expelidos durante a ejaculação. Os espermatozóides não podem passar do testículo para a ejaculação. No entanto, a função sexual é completamente preservada.
A infertilidade feminina é menos frequente. 80% das mulheres podem conceber e até dar à luz um recém-nascido saudável. A conceção é, no entanto, mais difícil devido ao muco espesso presente no colo do útero, que dificulta a passagem dos espermatozóides para o óvulo.
A gravidez em si também acarreta riscos, agravando o curso da doença e aumentando as necessidades de oxigénio e nutrientes, pelo que é necessário planear a gravidez com antecedência, ter em conta o estado de saúde da doente e acompanhar cuidadosamente toda a gravidez.
Em caso de problemas com a gravidez, pode recorrer-se à fertilização in vitro em centros de reprodução assistida.
Glândulas sudoríparas e fibrose quística
As pessoas com fibrose quística suam tal como as pessoas saudáveis e segregam a mesma quantidade de suor.
No entanto, a absorção de sais prejudicada significa que o NaCl não é reabsorvido no ducto sudoríparo, pelo que estes sais são subsequentemente excretados para a superfície da pele a uma taxa aumentada.
Esta caraterística de "pele salgada" foi considerada um sintoma de FC no passado. De facto, 1-2% dos doentes têm uma função normal do ducto sudoríparo.
Diagnóstico
Para além dos sintomas típicos da doença, há uma série de testes que podem confirmar o diagnóstico de fibrose quística.
Um grande avanço no diagnóstico foi a introdução do rastreio neonatal do bebé inteiro para confirmar a fibrose quística.
Este teste envolve a análise da tripsina imunorreactiva a partir de uma gota de sangue seca retirada do calcanhar do recém-nascido. Na FC, este parâmetro encontra-se elevado no sangue. Se for positivo, a criança é posteriormente submetida a um teste genético para confirmar a mutação genética do gene da FC.
Outro teste laboratorial é a chamada iontoforese de pilocarpina, que determina o teor de cloreto do suor. O nível normal de Cl é de até 40 mmol/l. Uma concentração superior a 60 mmol/l confirma uma perturbação do canal de cloreto.
O teste é considerado conclusivo se pelo menos três amostras forem positivas. O teste é efectuado em centros especializados.
As análises sanguíneas hematológicas e os testes de factores de hemocoagulação indicam uma perturbação hemorrágica em caso de lesão hepática grave.
A análise da perda de gordura, que é excretada nas fezes, mostra o grau de comprometimento da função pancreática.
Em particular, a função pulmonar é examinada em pormenor. Os exames importantes incluem uma radiografia do tórax, que mostra alterações no parênquima pulmonar.
Um outro exame funcional dos pulmões é a espirometria, em que o doente expira para um tubo e, com base nas medições do fluxo de ar, o médico avalia a capacidade funcional dos pulmões e, consequentemente, o grau de lesão do tecido pulmonar.
O exame microbiológico da expetoração tossida, das zaragatoas nasais ou das amígdalas pode revelar a presença de agentes patogénicos como Pseudomonas, S. Aureus, H. Influenzae ou Burkholderia cepacia, que são bactérias típicas que afectam o sistema respiratório dos doentes com FC.
O exame destes agentes patogénicos requer uma cultura demorada (crescimento de bactérias a partir de uma zaragatoa) e também um tratamento prolongado.
Para além da radiografia dos pulmões e do abdómen, também é utilizada a ecografia (USG) do fígado, da vesícula biliar ou do pâncreas ou a TCAR, que monitoriza a extensão das bronquiectasias e do enfisema pulmonar.
Se houver uma história familiar de fibrose quística, pode ser efectuado um aconselhamento genético antes de uma gravidez planeada. Atualmente, também é possível o diagnóstico pré-natal, ou seja, o teste genético da criança antes do nascimento.
Curso
As primeiras manifestações da FC surgem mais frequentemente na infância.
No entanto, nas últimas décadas, 30% dos doentes com FC foram diagnosticados na idade adulta.
Quando a infertilidade é investigada na idade adulta, pode ser encontrada uma função reduzida do canal de cloreto devido a uma mutação genética na fibrose quística.
A presença de FC na infância é indicada por sintomas típicos como a obstrução intestinal (íleo meconial) no recém-nascido imediatamente após o nascimento. Um segundo sintoma suspeito no recém-nascido é a duração prolongada da iterícia neonatal.
As crianças em idade pré-escolar sofrem frequentemente de pólipos nasais precoces (crescimentos da mucosa nasal) e de infecções sinusais recorrentes.
Nas crianças mais velhas, os sintomas mais graves já estão presentes, tais como uma função hepática afetada, enrijecimento e fibrose do fígado, tosse com muco sanguinolento ou uma maturação sexual afetada (puberdade atrasada).
Ao longo da vida, os doentes sofrem de inflamação recorrente das vias respiratórias, tanto do trato respiratório superior como dos pulmões.
A tosse é outro sintoma da FC, que se manifesta através da tosse com muco purulento contendo bactérias típicas da FC, nomeadamente Pseudomonas aeruginosa, Burkholderia cepacia e Haemophilus influenzae.
Com uma concentração insuficiente e prolongada de oxigénio no sangue, os vasos sanguíneos das pontas dos dedos começam a abrir-se e a alargar-se de forma compensatória, o que se designa por dedos em bico.
Com o passar do tempo, todas as funções dos órgãos afectados se deterioram, surgindo inflamações repetidas do pâncreas, diabetes, cólicas da vesícula biliar com cálculos biliares, doenças do fígado, fezes gordurosas e volumosas, carências vitamínicas com má nutrição.
Os doentes em idade reprodutiva sofrem de infertilidade, especialmente os homens. As mulheres conseguem engravidar, mas a gravidez é complicada tanto para a mãe como para o feto.
A sobrevivência dos doentes com FC aumentou significativamente devido à disponibilidade de tratamentos modernos. No passado, viviam no máximo 10-15 anos. Atualmente, os doentes vivem até aos 45 anos ou mais.
A causa de morte é, na maioria das vezes, insuficiência respiratória, infeção do trato respiratório com sépsis ou hemorragia de varizes esofágicas na cirrose hepática.
Como é tratado: título Fibrose cística
Tratamento da fibrose quística: medicamentos, inalação e terapia de suporte
Mostrar maisFibrose cística é tratado por
Outros nomes
Mucoviscidose
Recursos interessantes

MUDr. Andrea Bullová
Ver todos os artigos do mesmo autor