- solen.cz - Polirradiculoneurite de Guillain Barré, Doutor František Cibulčík, Departamento de Neurologia, SZU e UNB, Hospital Ružinov, Bratislava
- pubmed.ncbi.nlm.nih.gov - Síndrome de Guillain-Barré, Vibhuti Ansar, Nojan Valadi
- pubmed.ncbi.nlm.nih.gov - Compreender a síndrome de Guillain-Barré, Robert Estridge, Mariana Iskander
- pubmed.ncbi.nlm.nih.gov - Infecções por Campylobacter jejuni e citomegalovírus (CMV) em doentes com síndrome de Guillain-Barré, D Orlikowski, S Quijano-Roy, V Sivadon-Tardy, J-C Raphael, J-L Gaillard
O que é a síndrome de Guillain-Barré e quais são os seus sintomas e causas?
Fonte fotográfica: Getty images
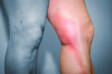
A Síndrome de Guillain-Barré (SGB) é uma doença desmielinizante inflamatória adquirida, denominada polirradiculoneuropatia, ou seja, o envolvimento de múltiplos nervos periféricos e raízes nervosas.
Sintomas mais comuns
- Dores musculares
- Espiritualidade
- Tensão arterial baixa
- Defesa
- Formigueiros
- Perturbações da deglutição
- Fraqueza muscular
- Retenção urinária - anúria/retenção
Mostrar mais sintomas ᐯ
Características
O nome síndrome de Guillain-Barré é uma designação mais antiga para o quadro de uma doença neurológica que se apresenta como uma paralisia flácida do corpo de início agudo.
A primeira referência a esta doença foi feita por Landry em 1859 e, mais tarde, em 1916, Guillain, Barré e Strohl aperfeiçoaram a descrição clínica dos sintomas e foram também os primeiros a descobrir os achados característicos do liquor.
Atualmente, utiliza-se a designação moderna de polirradiculopatia desmielinizante inflamatória aguda (PIDA).
Esta doença é mediada imunologicamente e envolve uma desregulação do sistema imunitário com uma propensão para reacções auto-imunes dirigidas contra os tecidos do sistema nervoso do próprio corpo.
As polirradiculoneuropatias podem ser classificadas como agudas ou crónicas de acordo com a evolução clínica.
Com base na constelação de sintomas e nos achados electrofisiológicos (por exame EMG), são classificadas numa das várias variantes de SGB.
Os seguintes subtipos pertencem ao grupo de doenças com um quadro de SGB:
- polirradiculopatia desmielinizante inflamatória aguda (AIDP)
- neuropatia axonal motora aguda (AMAN)
- neuropatia axonal motora e sensorial aguda (AMSAN)
- Síndrome de Miller-Fisher (MFS)
- neuropatia sensorial aguda e pandisautonomia aguda
O tipo mais comum de SGB é a polirradiculopatia desmielinizante inflamatória aguda (PIDA), na qual os sintomas máximos aparecem em poucos dias (máximo 4 semanas).
Segue-se uma estabilização dos sintomas, a chamada fase de planalto, após a qual o estado clínico melhora gradualmente.
No diagnóstico diferencial, é essencial distinguir a polirradiculoneuropatia desmielinizante inflamatória crónica (PDIC), uma doença mais lenta, progressiva ou com recaídas.
As variantes axonais da SGB podem ser puramente motoras ou neuropatias sensoriais e motoras combinadas. Ambas as variantes podem apresentar-se como doença muito grave com recuperação apenas parcial.
Com o declínio da poliomielite aguda (uma doença infecciosa viral que causa paralisia dos membros), a SGB tornou-se a doença paralítica aguda mais comum no mundo ocidental.
A incidência do SGB é de aproximadamente 1 a 2 casos por 100 000 habitantes por ano. Afecta mais homens do que mulheres, numa proporção de 3:2. É mais comum nos idosos, mas também pode ocorrer em indivíduos jovens.
A SGB está frequentemente associada à vacinação. Até à data, o aumento da incidência da doença só foi confirmado em associação com a vacinação contra a raiva. A vacina contra a raiva contém material do cérebro.
A probabilidade de ocorrência de SGB é de uma em cada 1000 pessoas vacinadas.
Compromissos
Até 80% dos doentes com SGB tinham uma doença infecciosa do trato respiratório (58%) ou gastrointestinal (22%) várias semanas antes do início dos sintomas, pelo que tem sido dada maior atenção à teoria pós-infecciosa da SGB.
Os agentes patogénicos infecciosos mais comuns são:
- Campylobacter - Provoca enterite por Campylobacter, uma doença do trato gastrointestinal. Os sintomas caracterizam-se por febre alta, dor de cabeça, dores abdominais com cólicas, especialmente no abdómen inferior direito, que podem simular apendicite. Está presente diarreia com sangue, com mistura de pus e muco. Os doentes têm náuseas, mas não vomitam.
- O micoplasma é uma bactéria sem parede celular que causa pneumonia atípica.
- Citomegalovírus - Um vírus que não causa sintomas em pessoas saudáveis. No entanto, em pessoas imunocomprometidas, por exemplo, após transplante de órgãos, a infeção é grave. Apresenta-se com febre, gânglios linfáticos aumentados, fraqueza, fadiga, dores musculares e articulares e inapetência. Os sintomas mais graves são hepatite, pneumonia, encefalite e infecções do esófago, do cólon e dos olhos.
- Vírus Epstein-Barr - O agente causador da mononucleose infecciosa. A doença é muitas vezes confundida com faringite estreptocócica devido aos sintomas semelhantes. O vírus afecta as glândulas endócrinas, o baço, o fígado, os gânglios linfáticos e a laringe.
- Haemophilus - A doença infecciosa mais grave é a epiglotite infecciosa, que também provoca sinusite, otite média, pneumonia e meningite purulenta grave.
Esta infeção é o estímulo para a produção de anticorpos. Com base na semelhança estrutural e química das moléculas do invólucro destes agentes patogénicos com os tecidos do próprio corpo, são produzidos anticorpos que são dirigidos contra as estruturas do próprio corpo.
Trata-se de um "engano" do inimigo com base na sua semelhança com o próprio organismo. Este fenómeno é designado em imunologia como mimetismo molecular.
A causa da SGB baseia-se em mecanismos auto-imunes de lesão dos nervos periféricos.
Nas formas desmielinizantes, os gangliosídeos das bainhas de mielina dos nervos são atacados por auto-anticorpos. Na forma axonal, os auto-anticorpos são dirigidos contra estruturas glicoproteicas localizadas na membrana celular dos processos nervosos chamados axónios.
A SGB recidiva com bastante frequência.
Os especialistas suspeitam, por isso, que determinados factores genéticos estejam envolvidos na doença.
Estes são os genes responsáveis pela ativação da imunidade e pela subsequente cascata de reacções auto-imunes.
Para além das doenças infecciosas e da predisposição genética, existem outros factores de risco para o desenvolvimento da SGB:
- hepatite anterior (inflamação do fígado)
- utilização de medicamentos como a heroína, a suramina e a estreptoquinase
- doenças crónicas, como o lúpus eritematoso sistémico e a infeção por VIH ou SIDA
- imunização ativa, ou seja, vacinação contra, por exemplo, a gripe, a raiva, etc.
Sintomas
Os sintomas descritos por Guillain, Barré e Stohl já em 1916 incluíam
- fraqueza muscular
- areflexia - ausência de reflexos tendino-musculares
- sintomas sensoriais como formigueiro e ardor com perda ligeira de sensibilidade
- dissociação albuminocitológica do líquido cefalorraquidiano (LCR)
Hoje em dia, já não se distinguem os diferentes sintomas consoante a variante da SGB que afecta o doente, existindo várias variantes clínicas, incluindo a neuropatia axonal motora aguda, a neuropatia axonal motora e sensorial aguda.
O sintoma comum a todas as variantes da SGB é a paralisia simétrica progressiva e a arreflexia ao longo de horas a dias. A paralisia progride de forma ascendente, ou seja, de baixo para cima, o que é muito típico da SGB. É acompanhada de dores musculares.
Primeiro, são afectadas as pernas, depois todos os membros inferiores. Mais tarde, surge a incapacidade de ficar de pé e de andar sobre os calcanhares. A progressão seguinte é a incapacidade de andar devido à fraqueza dos músculos das coxas, em particular. Gradualmente, a paralisia alastra aos membros superiores.
Se a doença não for tratada nesta fase, progride e o doente não consegue sentar-se, a sua face torna-se flácida devido à paralisia dos nervos e músculos faciais, não consegue levantar a cabeça, não consegue mover os olhos devido ao envolvimento dos nervos oculomotores.
Na fase mais grave da doença, a deglutição fica comprometida e o diafragma, o músculo respiratório mais importante, fica enfraquecido. O doente tem dificuldade em respirar, respirando apenas em respirações rápidas, ofegantes e curtas - taquipneia.
Ocorre hipoxia, ou seja, baixa concentração de oxigénio no sangue e nos tecidos, e as células do corpo começam a sufocar.
A insuficiência respiratória devido à insuficiência neuromuscular não é invulgar e exige o internamento na unidade de cuidados intensivos com necessidade de ventilação pulmonar artificial.
Simultaneamente, pode ou não ocorrer uma perturbação sensorial nas extremidades.
A par dos sintomas motores e sensoriais, desenvolvem-se sintomas autonómicos, que ocorrem em até 65% dos doentes internados, podendo ser muito graves e piorar o prognóstico global do doente.
Incluem, nomeadamente, os seguintes sintomas
- hipotensão ortostática (descida da tensão arterial quando se está de pé)
- anidrose (ausência de transpiração)
- retenção urinária
- atonia gastrointestinal (problemas de permeabilidade intestinal)
- iridoplegia (imobilidade das pupilas)
Uma variante da SGB, a síndrome de Miller-Fisher, que representa 5% dos casos de SGB, caracteriza-se pelos seguintes sintomas
- oftalmoplegia
- ataxia
- areflexia
A doença inicia-se com uma visão dupla (diplopia), seguida de uma perturbação da coordenação dos membros e da marcha.
Diagnóstico
Em 1981, foram estabelecidos os critérios de diagnóstico da SGB, há muito solicitados, que incluem fraqueza muscular progressiva em mais de dois membros, ausência de reflexos tendino-musculares nos membros e uma progressão que não dura mais de 4 semanas.
Os critérios de apoio incluem sintomas sensoriais ligeiros, simetria relativa dos sintomas, paralisia facial e um perfil albuminocitológico do líquido cefalorraquidiano.
Isto significa que existe uma concentração elevada de proteínas no líquido cefalorraquidiano, mas os resultados celulares são normais.
A única exceção a este critério são os doentes com infeção por VIH, em que este quadro no LCR é a norma.
Análise laboratorial do líquido cefalorraquidiano
Através de uma punção lombar, é possível obter o líquido cefalorraquidiano, que fornece informações valiosas sobre as reacções em curso no SNC, por exemplo, infecciosas ou auto-imunes.
Quando se suspeita de SGB, este exame é efectuado principalmente por razões de diagnóstico diferencial.
Um achado típico na SGB é o quadro da chamada dissociação proteico-citológica, ou seja, um aumento do conteúdo proteico na linfa com uma contagem baixa de células. No entanto, este achado ocorre apenas em 64 % dos doentes.
Os níveis elevados de proteínas durante os primeiros três dias ocorrem apenas em metade dos doentes e, após a primeira semana, em 80% dos doentes.
Este aumento dos níveis de proteínas também pode ser um falso positivo, que pode ser causado, por exemplo, pela administração de doses elevadas de imunoglobulinas no tratamento da SGB.
Uma contagem excessivamente elevada de células na linfa é um sinal de outro diagnóstico.
Doenças como tumores do palato mole, linfoma, radiculite por citomegalovírus, polineuropatia por VIH ou poliomielite - uma infeção causada por um vírus - são particularmente importantes no diagnóstico diferencial.
Exame eletrofisiológico (EMG)
Trata-se de um exame pormenorizado da condutividade dos nervos periféricos que, em neurologia, é um dos métodos mais comuns no diagnóstico de muitas doenças.
Fornece informações valiosas para o diagnóstico da SGB, especialmente para distinguir as suas variantes.
No entanto, mesmo este exame não fornece um resultado 100% seguro. Pode ocorrer confusão, por exemplo, numa fase muito precoce dos sintomas clínicos. Nessa altura, as condutividades nervosas medidas podem ser bastante normais.
Na maioria das vezes, a patologia é detectada no exame até duas semanas após o início dos sintomas, especialmente nos membros afectados.
As análises laboratoriais podem revelar um aumento da taxa de sedimentação de eritrócitos, resultados laboratoriais anormais dos parâmetros renais e hepáticos.
Estão também presentes perturbações de alguns electrólitos minerais, por exemplo, hiponatrémia (níveis baixos de sódio).
Na síndrome de Miller-Fisher, os anticorpos IgG séricos contra o gangliosídeo GQ1b estão presentes na maioria dos doentes.
Os anticorpos anti-GM1 e anti-GD1 (IgG) são frequentemente encontrados no sangue de doentes com a variante AMAN da SGB.
Curso
A SGB é uma doença relativamente aguda com um curso dramático e a necessidade de hospitalização com possível ligação a ventilação pulmonar artificial.
O início dos sintomas demora 4 semanas e o desaparecimento gradual de todos os sintomas pode demorar o dobro do tempo.
Em geral, os doentes afectados pela SGB têm um bom prognóstico. A modernização do tratamento e dos cuidados melhorou a sobrevivência dos doentes, tendo a mortalidade sido reduzida de 33% para 5-10%.
O maior avanço no tratamento da SGB foi a introdução da ventilação com pressão positiva.
A grande maioria dos doentes recupera da doença com apenas sequelas permanentes ligeiras no prazo de aproximadamente um ano. No entanto, alguns doentes sofrem danos irreversíveis e subsequente incapacidade neurológica permanente.
Aproximadamente 20% dos doentes apresentam paralisia permanente dos membros e atrofia muscular. As neuropatias sensoriais manifestadas por sensações desagradáveis, como formigueiros, formigueiros ou dormência, são incapacidades residuais comuns.
Muitos doentes referem também uma redução permanente do desempenho e fadiga crónica.
Como é tratado: título Síndrome de Guillain-Barré
Tratamento da síndrome de Guillain-Barré: medicamentos e terapia de apoio
Mostrar maisSíndrome de Guillain-Barré é tratado por
Outros nomes
Polirradiculopatia desmielinizante inflamatória aguda, AIDP, SGB
Recursos interessantes

MUDr. Andrea Bullová
Ver todos os artigos do mesmo autor